Required
Precursor m/z & Ion Mode
This defines the precursor ion mass-to-charge ratio (m/z).
This defines polarity and ensures correct model selection.
Example: PRECURSOR_MZ=517.22098
IONMODE=positiveIONMODE=negative
Comprehensive documentation for DeepMASS2 ncluding input format specifications, XCMS-based LC-MS/MS preprocessing, full annotation workflow, and result interpretation.
Before DeepMass annotation, raw LC-MS/MS data should be processed into DeepMass-compatible MSP or MGF files.
For large-scale annotation, use the local version.
These tags help DeepMASS2 parse spectra correctly, select the proper polarity-specific model, and assign meaningful output filenames.
This defines the precursor ion mass-to-charge ratio (m/z).
This defines polarity and ensures correct model selection.
Example: PRECURSOR_MZ=517.22098
IONMODE=positiveIONMODE=negative
Controls exported result filename and ion adduct. Defaults: [M+H]+ for positive, [M-H]- for negative.
DeepMASS2 uses this tag to define the output filename for the exported semantic similarity analysis.
If provided, the results will be saved as <COMPOUND_NAME>.csv.
Example: ADDUCT=[M-H]-
Example: COMPOUND_NAME=challenge_0
Adding the molecular formula helps the semantic similarity engine constrain potential chemical space, significantly improving the ranking accuracy of structurally related metabolites.
Example: FORMULA=C25H38O9
BEGIN IONS
PRECURSOR_MZ=194.04588176800002
COMPOUND_NAME=challenge_255
FORMULA=C9H9NO4
ADDUCT=[M-H]-
IONMODE=negative
132.0454 0.006008828855732822
134.0245 0.0022295306804225633
149.0483 0.00297960480724686
150.056 1.0
194.0459 0.36915136378467533
END IONS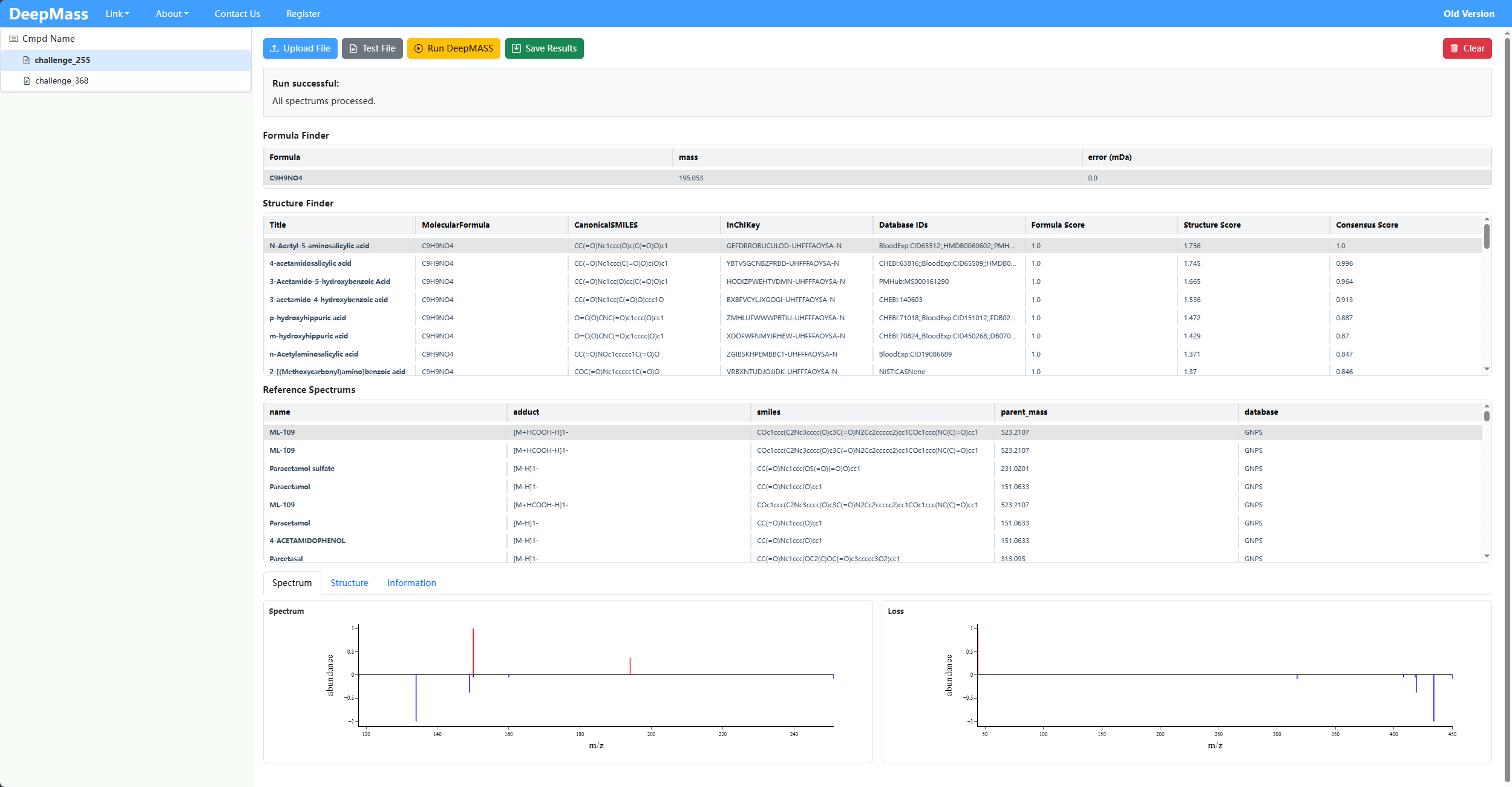
Annotates possible chemical formulas for unknown spectra.
C9H9NO4Predicts potential molecular structures with SMILES and structure scores.
Shows normalized intensity vs m/z, including unknown spectrum peaks and reference spectrum peaks for visual comparison.